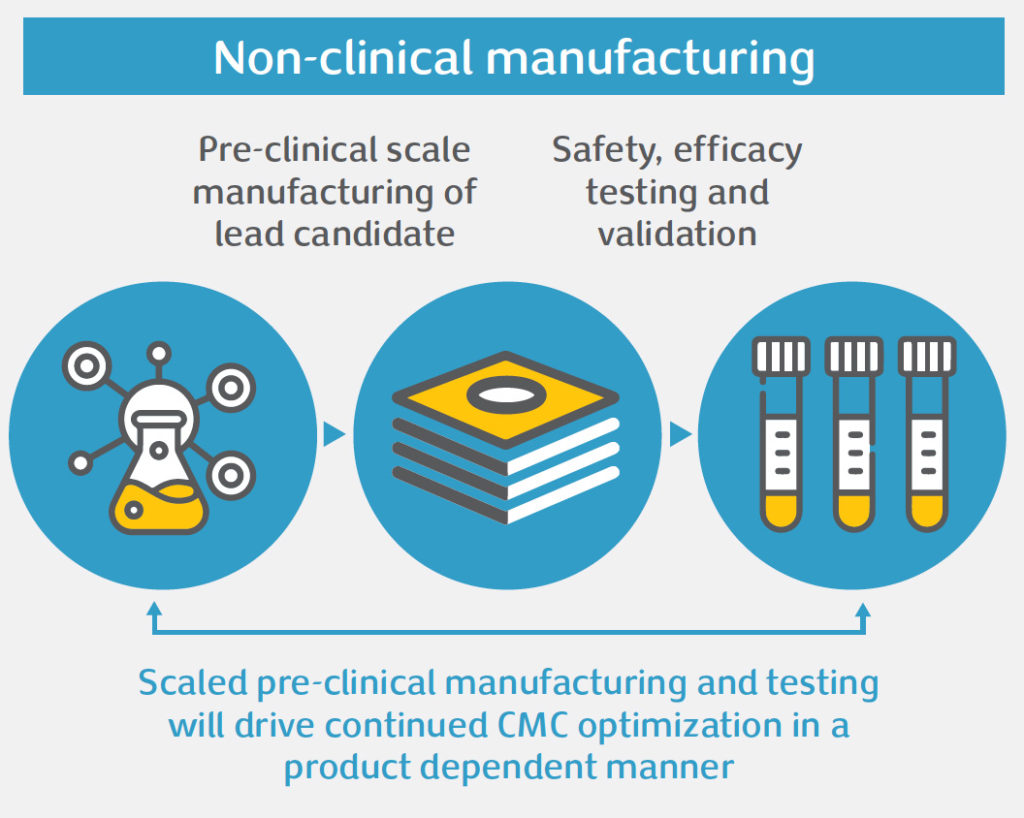
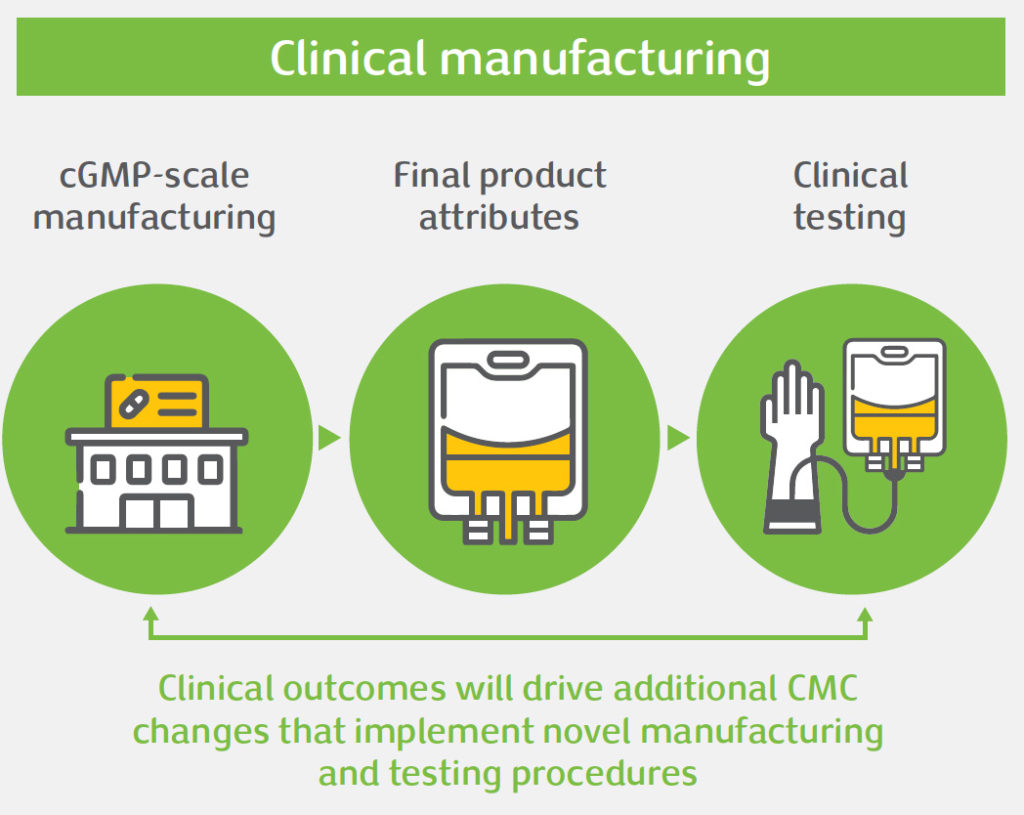
Gene therapy offers significant potential to treat diseases with high unmet medical need. However, the unique nature of these therapies poses challenges in product development, namely: safety concerns, efficacy issues, or obstacles related to Chemistry, Manufacturing and Controls (CMC). CMC is an integral aspect of gene therapy development. Done right, CMC development of complex manufacturing processes and analytical testing panels can accelerate clinical development and bring these important medicines to patients faster. For gene therapy products, CMC development often needs to be done in a highly compressed timeline, and there are important CMC challenges that need to be addressed to bring these products into the clinic and beyond, amongst which:

Gene therapy products are similar to other biological products in many ways. CMC hurdles in development of biologicals/ biotechnological products are common to our Venn experts; they have key experience in providing support in the development of a wide range of complex biological products. Their broad expertise allows them to have a holistic view on the CMC development of gene therapy products, and to provide out-of-the-box solutions. In example, their experience with human adenovirus-based vaccines allows to translate this expertise to adeno associated virus (AAV)- based gene therapies. The Venn CMC team is well aware of the requirements for early (Phase I) and later (Phase II and III) phases of development and can support you in CMC challenges you are facing in your gene therapy development. The Venn CMC team can help the client to find the optimal solutions to drive your product in development to clinical success.
One of the first steps is creating a solid roadmap for your product, a drug development plan, which outlines your drug development strategy and contains a carefully planned set of steps and action plans, progressing from non-clinical to the first-in-human Phase I to Phase II “proof of concept” and pivotal Phase III trials for registration. Our Venn experts can guide through your journey and can built a comprehensive drug development strategy for your product.
The next step is to support you in early and timely discussions with regulatory agencies on your development plan. Priority for agencies is to keep patients safe. Regulators want to see that a sponsor has control over its products and processes in development. Sponsors should therefore take advantage of, e.g., Innovation Task Force meetings and scientific advice meetings with the EMA or EU national authorities, and/ or INTERACT and Pre IND meetings with the US FDA, to seek advice and alignment with the agencies on CMC challenges you are facing, and the solutions and path forward you are proposing. The Venn regulatory team can plan and manage your health authority interactions. Our consultants are experienced in generating scientific advice/ Pre-IND briefing packages and liaising with US and EU regulatory agencies. They can also act as sponsor representative during EU scientific advice procedures.
Once you have tackled the CMC hurdles and you have buy-in from the agencies on your drug development plan, you can start planning the first-in-human Phase I trial for which you need to produce GMP-compliant investigational product, and for which you need to submit a clinical trial application. The Investigational Medicinal Product Dossier (IMPD) in the EU, or Investigational New Drug (IND) dossier in the US, is one of several pieces of the investigational product related data required for this application. The IMPD/ IND includes information related to the quality, manufacture, and control of the investigational product (including placebo). Guidance on the structure and CMC data requirements for investigational gene therapy products exists for EU and US applications. The EU and US guidelines for investigational
gene therapies have been built on the existing IMPD/ IND guidelines for biologicals/ biotechnological products, and the Venn CMC team is well equipped to support you in authoring the IMPD/ IND.
In addition to the clinical trial application, gene therapy products classified as genetically modified organisms will be subject to a GMO application to the applicable biosafety agencies in the EU to permit the release of the product into the environment (so-called biosafety review). Pending on the modalities of the use, the product can be considered to be contained use or deliberate release, each coming with a different set of requirements. The expectations for environmental risk assessments and GMO applications are regulated on a national level and the requirements and classification can therefore vary significantly between member states in the EU, resulting in a maze of requirements that are difficult to navigate. The Venn regulatory team can guide you in this process and can help you with the procedural aspects of the GMO application and clinical trial application.
In conclusion, there are important CMC challenges that need to be addressed to bring a gene therapy product into the clinic and beyond, but many aspects of gene therapy development are similar to other biological products. With guidance and expertise of our Venn experts in the biological/ biotechnological field, your chances of a successful outcome of your gene therapy development will be increased.



